 Allgemeine Informationen
Allgemeine Informationen General
information
General
informationPatient registries on deafness
What is a patient registry?
Registries are structured tools that support patient engagement in research for those who may otherwise face hurdles in connecting with rare disease studies. They are developed by researchers who are experts in a particular disorder as a way to collect valuable information that can be used to increase understanding of the disorder. Registries support research into pathogenesis, therapy, and longitudinal natural history studies and can serve as a way to recruit participants into clinical studies.
With respect to the Otoferlin and CABP2 registries, we would like to understand the genetic mutations in the OTOF and CABP2 genes and how these affect hearing over time, also in relation to hearing rehabilitation (hearing aids, cochlear implants, future gene therapies) and changes in body temperature (temperature-dependent hearing loss has been observed for some otoferlin mutations) as a way to continue to inform our research on the genetic causes of hearing loss.
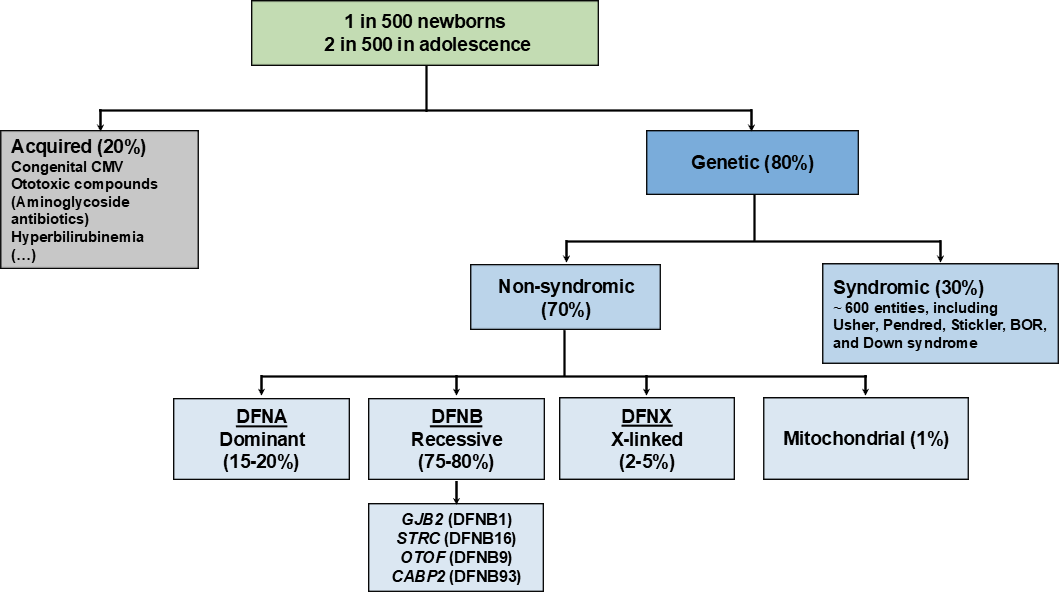
How frequent is deafness? What kinds of deafness are there?
Screening for hearing loss starts as part of universal newborn hearing screening in many parts of the world. These data already suggest that roughly 1 in 500 newborns have a hearing loss requiring treatmnet. We know that this roughly doubles in adolescents and steadily increases throughout the lifespan (Figure 1). It is important to highlight that hearing loss tends to be under-diagnosed and people can sometimes go many years before hearing loss is clinically diagnosed. Therefore, even in instances of suspected hearing loss, evaluation and testing for hearing loss by audiologists, otolaryngologists, and otologists is very important.
We can describe hearing loss and deafness according to several different parameters. For example, whether it is thought to be due to genetic or environmental causes. Early onset hearing loss is thought to be predominantly due to genetic causes. Among the inherited forms of hearing loss, 70% occur as isolated hearing loss, also termed non-syndromic hearing loss, whereas around 30% appear as syndromic hearing loss with additional features involving other organ systems. Among the isolated forms of hearing loss, 75-80% appear as an autosomal recessive form, which means that both copies of a gene inherited from the mother and father each have a mutation that is transmitted to their child. A unique feature about autosomal recessive forms of hearing loss is that there is usually no previous family history and the parents usually hear normally. Mutations affecting the genes GJB2 (DFNB1) and STRC (DFNB16) are the most common genetic causes for early onset hearing loss. OTOF (DFNB9) is also an important gene involved in hereditary hearing loss and a target for potential gene therapies (Figure 1). Hearing loss can also be transmitted in an autosomal dominant inheritance pattern, where mutations in a single copy of the affected gene can cause hearing loss. Here, there is usually a traceable family history with at least one parent affected with a similar course of hearing loss (such as onset and severity) as well. This is thought to be present in 15-20% of early onset hearing loss. The rarer forms of hereditary hearing loss include X-linked, mainly affecting males due to mutations in genes residing on the X chromosome (2-5%), and mitochondrial (~1%) (Figure 1).
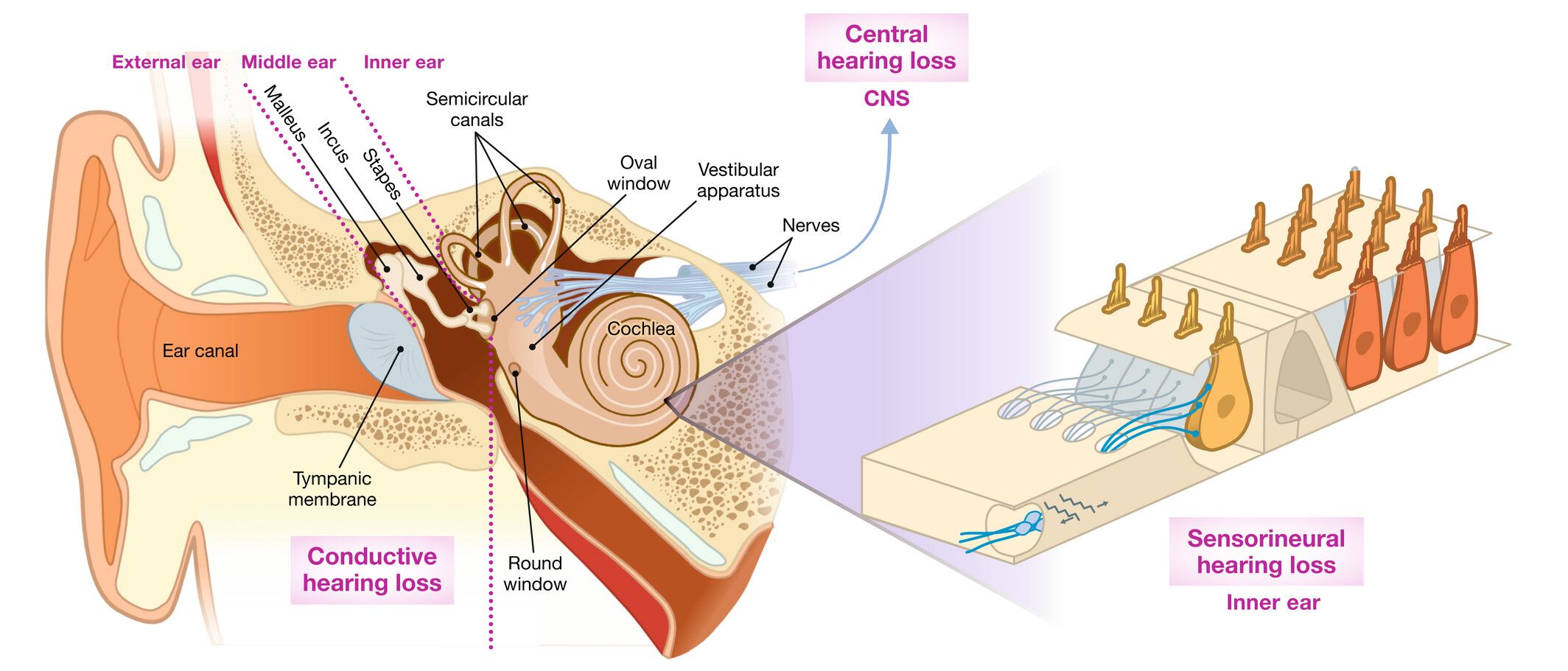
Hearing loss can also be described according to the precise location of the defect in the auditory system. For example, damage to the outer ear or middle ear can cause conductive hearing loss, whereas damage to the delicate cells of the cochlea and auditory nerve can cause sensorineural hearing loss. Defects at the synapse of the sensory receptor cells of the inner ear, the so-called hair cells, causes synaptopathy whereas damage to the spiral ganglion neurons forming the auditory nerve causes neuropathy. A combination of damage to both the outer, middle and inner ear can cause mixed hearing loss. Finally, damage to the central auditory pathway or auditory centre of the brain causes central hearing loss (Figure 2).
Molecular genetic testing can help to provide information about not only which cells in the auditory system that we should anticipate to be the origin of hearing loss but also about which proteins should be affected and which cellular functions should be involved. This can yield a greater gene-based understanding about other features related to the onset of hearing loss, such as severity and which frequencies will be affected, whether or not hearing loss will change or progress over time, whether both ears are expected to be equally affected, and whether or not a vestibular dysfunction may be possible (Figure 3).

How can deafness be treated?
Treatment of hearing loss involves close cooperation between ENT doctors and audiologists and is determined after a definite diagnosis. Hearing aids, amplifying specific frequencies that are deficient, are often helpful for individuals with a significant amount of “residual hearing” or remaining hearing function. Individuals with a more severe or profound degree of hearing loss are often candidates for cochlear implants that work to electrically stimulate the auditory nerve, bypassing the damaged site of the inner ear. These remain as the most effective means to treat hearing loss. Important research continues to further improve these treatment options (see also "Hearing the Light").
From pre-clinical studies, targeted gene therapies have emerged as promising ways to address the precise genetic cause for hearing loss but are not yet approved. We have made important contributions to understanding otoferlin-related deafness and continue to advance pre-clinical work. We would like to use this opportunity to use the registry as a tool for engaging otoferlin patients to gain further insight into otoferlin-related deafness to prepare future gene therapies.
Further Information
Further information can be found in this publication (from the journal HNO). Please note that this is a translation of a German article, which might not be completely accurate.